In this article, I will talk about the biochemistry of a disease that maybe many people have never heard of, marfan syndrome. We will learn soon the answers to such questions as "What is the cause, what are the symptoms?". But first I want to give some information. Abraham Lincoln, whom we all know, has this disease. He already shows that he carries this disease with his physical characteristics. Michael Phelps was also seen carrying Marfan syndrome more recently. This issue has been suspected but the tests carried out have proven that he does not carry the disease, even if he is in the risk group. That’s enough, now let's get to know the disease.
Marfan Syndrome is an autosomal dominant disorder on the chromosome 15 linked to the FBN-1 gene. It affects many things like skeleton, lungs, eyes, heart and blood vessels. It is characterized by an abnormal length of arms and legs. Well, what is this FBN-1 or fibrin? What does it do?
FBN1 encodes a protein called fibrillin. It is the major component of microfibrils that form the elastic fibers of the extracellular matrix in connective tissue with elastin.
Fibrillin is a group of large, disulfide-rich molecules dominated by calcium-binding EGF-like (cBEGF) dominantly found in connective tissue matrices, a glycoprotein. It contains 15% of cysteine amino acid. The two cysteines come together to form the disulfide bridge by turning into the cysteine.
Fibrillin monomers aggregate as microfibrils on the cell surface and are rapidly cross-linked to the extracellular matrix. Here they contribute to the elastic properties of tissues such as blood vessels. It holds the elastin in bundles. It is the basic component in the peripheral microfibrils of elastic fibers. Fibrillin-1 is a fibrillar protein that can also be found in elastic connective tissue and other connective tissue. This protein is required for the construction of elastic fibrils in the connective tissue. Many tissues would weak without the structural support provided by fibrillin. This would have serious consequences. For example, tears form on the walls of large arteries.
Fibrillin-1 (FBN1) mutations associated with Marfan syndrome cause pathogenic changes, including aortic dilatation and dissection, leading to an increase in transforming growth factor beta (TGF-Beta) activation in the connective tissue. In addition, the increase in TGF-Beta causes problems in the connective tissue of the body.
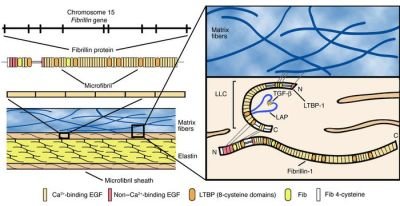
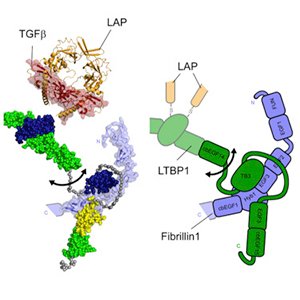
TGF-beta is secreted in the extracellular medium as a complex (LLC) covalently bound to the latent TGF-beta binding protein (LTBP). LTBPs interact with FBN1 through a characteristic 8-Cys module shared between two proteins.
It inhibits uncontrolled activation of TGF-Beta by binding as a microfibrillar regulator of mature TGF-Beta. TGF-Beta is inactivated by the controlled binding of LTBP to FBN1 under physiological conditions. Disruption of microfibrils is induced by insufficient or dysfunctional FBN1 leading to reduced binding capacity of LTBP and increased activity of TGF-Beta in extracellular medium. TGF-Beta causes collagen production. The ECM tightly regulates remodeling, leading to tissue fibrosis and endangering organ structure and function.
What Are The Observed Symptoms?
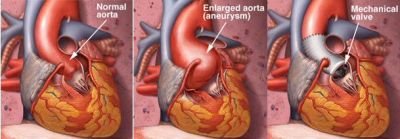
The most serious symptoms are due to cardiovascular system abnormalities such as decreased vascular smooth muscle cells (VSMC) and the integrity of the tunica media area of the descending aorta, which is composed of elastic fibers. These aortic abnormalities are due to elastic fiber breakdown and increased collagen deposition. Severe effects of the mutation are usually asymptomatic until a life-threatening aortic aneurysm or dissection occurs.
Symptoms of severe aortic regurgitation (dyspnoea, orthopnea, paroxysmal dyspnea, excessive sweating) are seen with the enlargement of the aortic ring. Myocardial hypertrophy, anginal pain, heart failure at a later age, pulmonary edema may develop.
Mitral and tricuspid valves often show loss of elasticity or inadequacy in patients with Marfan syndrome. Ocular complications can be early cataracts or glaucoma. Patients with Marfan syndrome, pneumothorax including pectus carinatum and excavatum (extinction due to air in the chest cavity) and anterior chest deformities may cause restrictive lung disease. People with Marfan syndrome are often quite long and weak. Arms, legs, fingers and toes are disproportionate and may look too long compared to the rest of the body.
How to Diagnose?
Mutation studies on the FBN1 gene of patients affected by the disease have revealed that almost all families have different FBN1 mutations. Therefore, linkage analyzes must be performed on families where genetic testing should be applied. 15-30% of the mutations are caused by new mutations.
Clinical features and family history are important when Marfan syndrome is diagnosed because there are different disorders like Marfan with fibrillin gene mutation. Some mental disorders accompanying Marfan syndrome have also been reported in the literature. Some case studies have suggested that Marfan syndrome may be caused by psychiatric disorders as well as systemic diseases. Concomitant mental disorders with connective tissue diseases are not directly explained. However, it has previously been hypothesized that defects in microfibril genes such as fibrillin may be predisposed to neurodevelopmental anomalies.
What Are The Treatment Approaches?
The damaged vessel is replaced by an artificial vein attached to a 10 cm cadaver against tears that can form on the arterial walls. But as a result, one has to use pills that prevent clotting throughout life. Sometimes beta-blockers are used to lower high blood pressure. Administration of losartan reduces TGF- β production and increases proinflammatory response, myofibroblast differentiation and reactive oxygen species. Orthopedic treatment helps remove the effects of pectus carinatum and excavatum.
Studies
Inhibition of TGF-β using naturally neutralizing antibodies or indirectly using drugs such as Losartan showed the reversal of pathological changes in aorta, lung, and skeleton in FBN1 mouse models and suggested that FBN1 levels in ECM affected TGF-β activity.
Subsequent studies have shown that the changes in the mechanization of both the integrin levels and the angiotensin receptor can further modulate conditional FBN knockout mouse phenotypes and display a complex cell/matrix interaction.
References
- Hayward, C., Brock, D.J.. Fibrillin-1 mutations in Marfan syndrome and other type-1 fibrillinopathies.1997
- Dietz, H. C., Pyeritz, R. E. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders.Human Molecular Genetics. 4/1799–1809. 1995
- Robertson, I. B., Dias, H. F., Osuch, I. H., Lowe, E. D., Jensen, S. A., Redfield, C., Handford, P. A., The N-Terminal Region of Fibrillin-1 Mediates a Bipartite Interaction with LTBP1. Structure. 8,1208–1221
- https://www.jci.org/articles/view/22399
- http://www.marfansresearchfoundation.ie/update-on-research/
Being A SteemStem Member
Interesting Article But Some Words Are Too Difficult To Understand :D
Will Study Two Three Times More To Understand It :D
kedi, I see that you are a chemical engineering student. Where did you get the idea to make this post? Do you know anyone with Marfan's? Do you work on any project involved with this disease? What's your connection to Marfan's and why the interest?
Recently I have seen related articles in some journals. I was interested and have done a little more research. So I have not been on any project related to this disease yet. Just wondering.